
Philosophy of SCINE Heron
SCINE Heron is the graphical user interface for all other SCINE modules. It has six ways
operation.
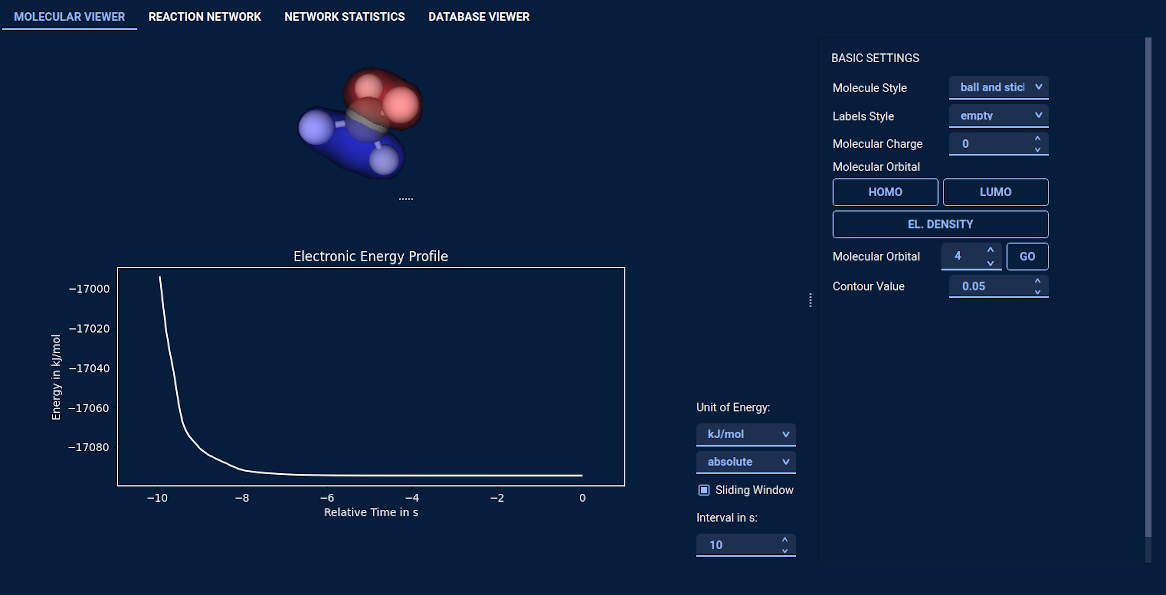
1) One can explore chemical reactivity immersively and interactively based on first principles in real-time. The
graphical user interface of SCINE Heron displays a three-dimensional molecular structure and allows users
to interact with it. They can induce structural changes with a computer mouse or a haptic device and perceive
the effect of their manipulations immediately through visual and/or haptic feedback. For fast electronic
structure calculations, we employ currently different semi-empirical methods, which can deliver properties
in the millisecond timescale, while providing a qualitatively correct description of the potential energy surface.
2) One can construct a system-focused atomistic model (SFAM), i.e., a force field parametrized on Hessian calculations
on the specific system. SFAM force fields can be employed in the interactive setting either as a stand-alone or
in a hybrid QM/MM model. The hybrid model can also be constructed within the graphical user interface either by our
automated QM region selection algorithm or by manual selection of the QM region by clicking on the QM atoms.
3) One can launch individual energy calculations and optimization routines, such as transition state searches,
with any backend program supported in SCINE, in the ReaDuct tab. The input structures can be read-in from file
or stem directly from Interactive or an explored reaction network.
4) One can automaticaly determine the active space for multi-configurational calculations with autoCAS.
Based on orbital entanglement measures derived from an approximate DMRG wave function, autoCAS identifies all strongly
correlated orbitals to be included in the active space of a final, converged calculation.
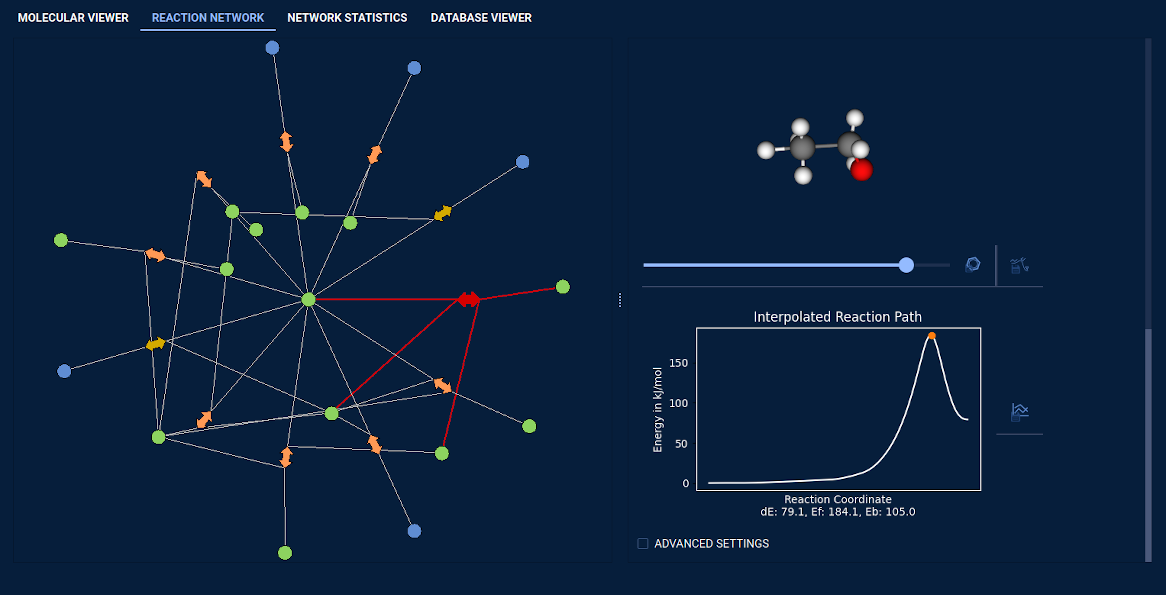
5) One can interact with explorations done by SCINE Chemoton. One can visualize the chemical reaction
network without drowning in too much information. For example, one can selectively display reactions with a
barrier lower than a certain, user-specified threshold. Furthermore, one can analyze all compounds and reactions
discovered, e.g., for reactions, one can visualize the trajectory and study the energy along it. It is also possible
to search and visualize all pathways between two given compounds. Any compound discovered by Chemoton can be
transferred to the interactive part of the GUI, allowing it to be further studied.
6) One can carry out reaction explorations directly in the graphical user interface by either constructing individual
Chemoton engines, or by relying on the Steering Wheel mechanism that lets one guide the automated exploration more closely.
The exploration can be constrained by additional aggregate and reactive site filters that can also be constructed in the
graphical user interface. Almost all of the exploration guidances in Heron support I/O operations which allow to save
and reuse individual parts for other explorations or restarts.
Technical Details
Heron is implemented in Python. It can be installed either from source or via PyPI. To work properly, Heron needs a few other SCINE modules, such as:
- SCINE Database, which is necessary to access a database containing an exploration done with Chemoton,
- SCINE Sparrow, which allows to do semiempirial calculations, in an interactive fashion, and
- SCINE Utilities, which is a library of common functionality used across all SCINE modules.
Current Features
- Interactive Molecular Viewer
- Real-time calculations of energies and forces (using SCINE Sparrow)
- Haptic device support
- Real-time energy plot
- Basic molecular building/editing
- Isosurface plots of orbitals and densities
- Reaction Network Viewer
- Excerpt view of compounds and reactions
- Different filtering options
- Navigation around a single centered Compound
- (Shortest) path searches based on Compound IDs with Pathfinder
- Expansion tab for Compounds (showing contained Structures)
- Expansion tab for Reactions (showing contained Elementary Steps)
- SVG export of all graph views
- SCINE Database Statistics
- Database content statistics
- Calculation status statistics
- Runtime histogram
- SCINE Database Browser
- Listing, searching and displaying of individual database entries
- Reaction and Elementary Steps
- Compounds and single-molecule Structures
- Flasks and multi-molecule complexes (also Structures)
- SCINE Chemoton
- Set up, start, stop, and interact with explorations
- Interactively carry out exploration by means of the Steering Wheel approach
- SCINE autoCAS
- Determine active spaces for multireference calculations
Download
SCINE Heron is distributed as an open source code. Visit our GitHub page to download it.
Future Releases
- Improve user experience
Documentation
Documentation is provided in the source code; you can also access it here.
Support
Despite intense testing of the program, questions may arise with respect to the usage of SCINE Heron. Do not hesitate to contact the developers via scine@phys.chem.ethz.ch in case of any questions and suggestions.
References
-
Primary reference for Heron 2.0.0:
M. Bensberg, G. P. Brandino, Y. Can, K.-S. Csizi, M. Del, S. A. Grimmel, M. Mesiti, M. Mörchen, C. H. Müller, M. Steiner, P. L. Türtscher, J. P. Unsleber, M. Weberndorfer, T. Weymuth, M. Reiher, "qcscine/heron: Release 2.0.0 (Version 2.0.0)", Zenodo, 2024. DOI - K. H. Marti, M. Reiher, "Haptic quantum chemistry", J. Comput. Chem., 2009, 30, 2010. DOI
- M. P. Haag, M. Reiher, "Real‐time quantum chemistry", Int. J. Quantum Chem., 2013, 113, 8. DOI
- M. P. Haag, M. Reiher, "Studying chemical reactivity in a virtual environment", Faraday Discuss., 2014, 169, 89. DOI
- M. P. Haag, A. C. Vaucher, M. Bosson, S. Redon, M. Reiher, "Interactive Chemical Reactivity Exploration", ChemPhysChem, 2014, 15, 3301. DOI
- A. H. Mühlbach, A. C. Vaucher, M. Reiher, "Accelerating Wave Function Convergence in Interactive Quantum Chemical Reactivity Studies", J. Chem. Theory Comput., 2016, 12, 1228. DOI
- A. C. Vaucher, M. P. Haag, M. Reiher, "Real‐time feedback from iterative electronic structure calculations", J. Comput. Chem., 2016, 37, 805. DOI
- A. C. Vaucher, M. Reiher, "Molecular Propensity as a Driver for Explorative Reactivity Studies", J. Chem. Inf. Model., 2016, 56, 1470. DOI
- A. C. Vaucher, M. Reiher, "Steering Orbital Optimization out of Local Minima and Saddle Points Toward Lower Energy", J. Chem. Theory Comput., 2017, 13, 1219. DOI
- M. A. Heuer, A. C. Vaucher, M. P. Haag, M. Reiher, "Integrated Reaction Path Processing from Sampled Structure Sequences", J. Chem. Theory Comput., 2018, 14, 2052. DOI