QCMaquis
Philosophy of SCINE QCMaquis
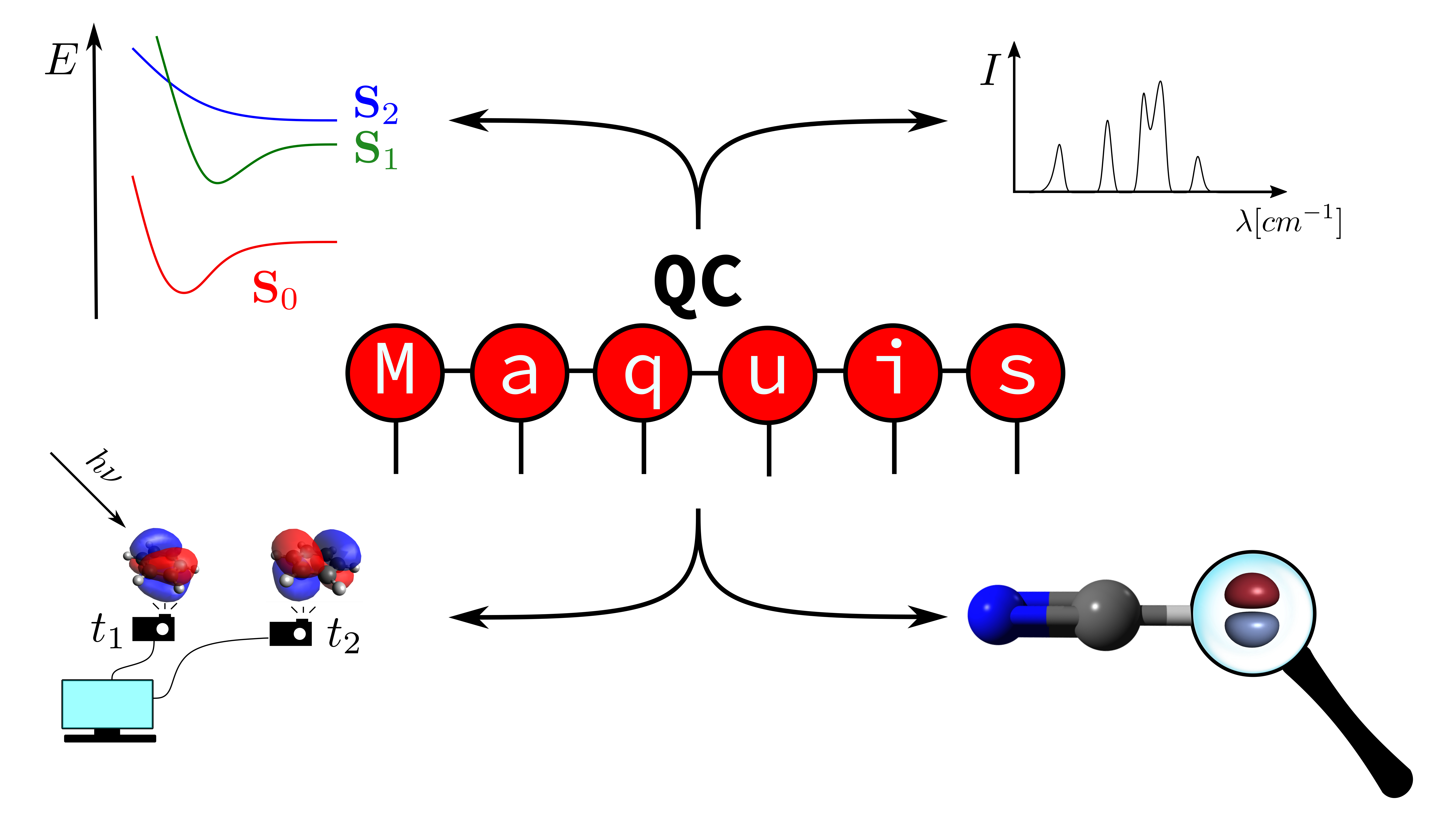
QCMaquis is a SCINE module devoted to exact molecular simulations based on the Density Matrix Renormalization Group (DMRG) algorithm. The DMRG method is a tensor-based algorithm for optimizing exact many-body wave functions of large quantum systems. QCMaquis offers a computational platform to work with multiple kinds of DMRG simulations, including
- the calculation of the electronic energy of molecular systems,
- the solution of the vibrational Schrödinger equation on anharmonic potential energy surfaces,
- the quantum dynamics simulation of non-equilibrium processes happening on time-scales ranging from the attosecond to the picosecond, and
- the calculation of accurate molecular energies without relying on the Born-Oppenheimer approximation.
Technical Details
QCMaquis is written in C++ (C++14 standard) and parallelizes the computationally heavy steps of a DMRG simulation with shared-memory parallelism (OpenMP). QCMaquis can be used via command-line tools, as a standalone software, or used in combination with the OpenMolcas software for quantum chemical simulations. We encourage to install QCMaquis via the OpenMolcas software for electronic-structure calculation, and use the code as a standalone software for simulations beyond time-independent electronic-structure calculations. Note that QCMaquis can be interfaced with the SCINE autoCAS module to analyze the results of a DMRG simulation based on orbital entanglement measures.
Current Features
- Calculation of full configuration interaction and complete active space configuration interaction ground-state electronic energies with DMRG
- Spin-adapted DMRG formulation for targeting wave functions belonging to specific spin representations
- State-specific optimization of excited-states with DMRG
- Calculation of one-, two-, three-, and four-particle reduced density matrices
- Variational inclusion of relativistic effects based on the four-component relativistic electronic Hamiltonian
- Calculation of one- and two-orbital entanglement measures
- Interface to the OpenMolcas program package for:
- DMRG-SCF calculations with and without the inclusion of environmental effects (e.g. PCM)
- State-specific and state-averaged DMRG-SCF calculations
- Analytic gradients for state-specific DMRG-SCF calculations
- Calculation of spin-orbit coupling matrix elements, electronic and magnetic properties with the state-interaction approach
- State-specific and quasi-degenerate (multi-state) DMRG-NEVPT2 calculations
- Simulation of non-equilibrium electron dynamics driven by the non-relativistic electronic Hamiltonian
- Optimization of ground-state wave functions with the imaginary-time time-dependent DMRG method
- Calculation of molecular energies with pre--Born--Oppenheimer, nuclear-electron Hamiltonians
- Support for the vibrational DMRG method for large-scale anharmonic vibrational calculations
- Advanced state-specific excited-state targeting for high-energy excited states
Download
SCINE QCMaquis is distributed as an open source code. Visit our GitHub page to download it. Visit the OpenMolcas GitHub page for more information on the QCMaquis/OpenMolcas interface.
Future Releases
- Simulation of femtosecond processes with vibronic Hamiltonians
- Non-equilibrium vibrational and electronic dynamics driven by time-dependent perturbations
- Explicitly-correlated DMRG calculations with the transcorrelated Hamiltonian
- Python bindings for all the functionalities offered by QCMaquis
- Distributed memory parallelization with MPI
Documentation
A detailed installation guide and manual for QCMaquis (and its components) can be found in the "doc" folder in the source code.
Support
Do not hesitate to contact the developers via dmrg@phys.chem.ethz.ch not only in case of any questions that arise, but also for feature suggestions and bug reports. The latter will be greatly appreciated.
References
For reproducibility reasons, please cite, depending on the actual calculations you carried out, one or more of the following papers in publications that present data produced with QCMaquis:
-
Overview on DMRG-based methods for molecular simulations:
A. Baiardi, M. Reiher, "The density matrix renormalization group in chemistry and molecular physics: Recent developments and new challenges" J. Chem. Phys., 2020, 152, 040903. DOI -
Non-relativistic electronic structure calculations:
- S. Keller, M. Dolfi, M. Troyer, M. Reiher, "An efficient matrix product operator representation of the quantum chemical Hamiltonian", J. Chem. Phys., 2015, 143, 244118. DOI
- Spin-adapted DMRG: S. Keller, M. Reiher, "Spin-adapted matrix product states and operators", J. Chem. Phys., 2016, 144, 134101. DOI
- Details of the DMRG-SCF method: Y. Ma, S. Knecht, S. Keller, M. Reiher, "Second-Order Self-Consistent-Field Density-Matrix Renormalization Group", J. Chem. Theory Comput., 2017, 13, 2533. DOI
- DMRG-NEVPT2 method: L. Freitag, S. Knecht, C. Angeli, M. Reiher, "Multireference Perturbation Theory with Cholesky Decomposition for the Density Matrix Renormalization Group", J. Chem. Theory Comput., 2017, 13, 451. DOI
- State interaction method: S. Knecht, S. Keller, J. Autschbach, M. Reiher, "A Nonorthogonal State-Interaction Approach for Matrix Product State Wave Functions", J. Chem. Theory Comput., 2016, 12, 5881. DOI
-
Vibrational DMRG:
- A. Baiardi, C. J. Stein, V. Barone M. Reiher, "Vibrational Density Matrix Renormalization Group", J. Chem. Theory Comput., 2017, 13, 3764. DOI
- A. Baiardi, C. J. Stein, V. Barone M. Reiher, "Optimization of highly excited matrix product states with an application to vibrational spectroscopy", J. Chem. Phys., 2019, 150, 094113. DOI
-
Relativistic electronic structure calculations:
S. Battaglia, S. Keller, S. Knecht, "Efficient Relativistic Density-Matrix Renormalization Group Implementation in a Matrix-Product Formulation", J. Chem. Theory Comput., 2018, 14, 2353. DOI -
Time-dependent DMRG:
- Theoretical foundations: A. Baiardi, M. Reiher, "Large-Scale Quantum Dynamics with Matrix Product States", J. Chem. Theory Comput., 2019, 15, 3481. DOI
- Non-equilibrium electron dynamics with DMRG: A. Baiardi, "Electron Dynamics with the Time-Dependent Density Matrix Renormalization Group", J. Chem. Theory Comput., 2021, 17, 3320. DOI
-
Nuclear-electron DMRG:
- A. Muolo, A. Baiardi, R. Feldmann, M. Reiher, "Nuclear-electronic all-particle density matrix renormalization group", J. Chem. Phys., 2020, 152, 204103. DOI
- R. Feldmann, A. Muolo, A. Baiardi, M. Reiher, "Quantum Proton Effects from Density Matrix Renormalization Group Calculations", J. Chem. Theory Comput., 2022, 18, 234. DOI