Molassembler
Philosophy of SCINE Molassembler
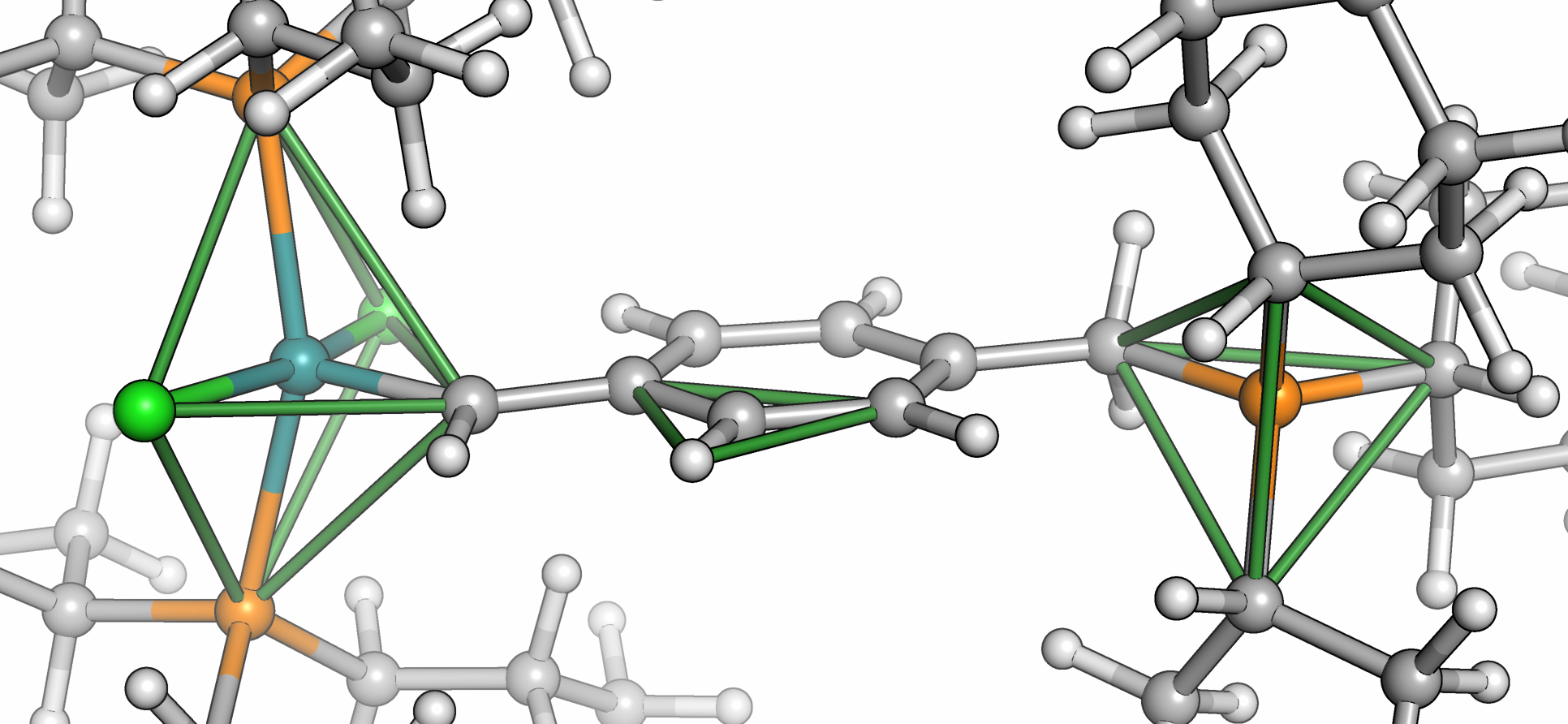
Molassembler is a C++ library that aims to facilitate conversions between Cartesian and graph representations of molecules. It provides the necessary functionality to represent a molecule as a graph, modify it in graph space, and generate coordinates from graphs. It can capture the absolute configuration of molecules with multidentate and haptic ligands from positional data and generate non-superposable stereopermutations as output. Its molecular model is split into a graph and a list of objects named stereopermutators managing the relative spatial orientation of an atom's or bond's substituents. Atom stereopermutators manage this arrangement in distinct polyhedral shapes that range from two substituents (linear and bent) up to twelve (icosahedron and cuboctahedron). Bond stereopermutators enumerate the rotational alignments of two adjacent atom stereopermutators.
Technical Details
Molassembler is a C++17 library and Python bindings. Core algorithms such as shape classification from Cartesian coordinates and conformer generation are trivially parallelized. Molassembler makes heavy use of the private implementation pattern to separate implementation details from the general API. The library itself draws from multiple sub-libraries each serving separate purposes.
Current Features
- Molecules can be constructed from many types of information.
- Stereocenters are treated from trigonal pyramidal all the way up to icosahedral and cuboctahedral local shapes.
- A high-temperature approximation is invoked by default to avoid considering inverting nitrogen centers as stereocenters, but this is optional. Even in the high-temperature approximation, nitrogen centers whose substituents form a strained cycle and hence do not invert rapidly are considered a stereocenter.
- All stereocenter permutations are generated with relative statistical occurrence weights. Linking of ligands (denticity) is properly considered. Several classes of haptic ligands are fully supported.
- Editing of molecules preserves chiral information by default, and is highly configurable.
- Molecules can be canonicalized for fast isomorphism tests. Canonicalization can be customized to use subsets of the available information for vertex coloring if desired.
- Ranking algorithms are nearly fully IUPAC Blue Book 2013 compliant, generalized to any local shape.
- Stochastic conformer generation with Distance Geometry
- Directed conformer generation through enumeration of rotamers
- Experimental SMILES parser notably implementing stereoconfiguration for square, trigonal bipyramid and octahedron shapes in addition to the tetrahedron.
Download
SCINE Molassembler is open source. Visit our GitHub page to download it.
Future Releases
- Better handling of aromaticity
- Better ranking
- Stabilized SMILES parser
Documentation
See the manual for detailed instructions on installation and introductory examples. A full documentation of the library API is available separately for the C++ library and its Python bindings module.
Support
A cheminformatics toolkit can always endeavor to be better or to expand its scope. Please open GitHub issues for bugs, and do not hesitate to contact the developers via scine@phys.chem.ethz.ch in case of questions and suggestions.
References
-
Introductory paper
J.-G. Sobez, M. Reiher, "Molassembler: Molecular graph construction, modification and conformer generation for inorganic and organic molecules", J. Chem. Inf. Model., 2020. DOI -
Primary reference for Molassembler 3.0.1:
M. Bensberg, S. A. Grimmel, J.-G. Sobez, M. Steiner, J. P. Unsleber, T. Weymuth, M. Reiher, "qcscine/molassembler: Release 3.0.1 (Version 3.0.1)", Zenodo, 2025. DOI