|
Scine::Sparrow
5.1.0
Library for fast and agile quantum chemical calculations with semiempirical methods.
|
|
Scine::Sparrow
5.1.0
Library for fast and agile quantum chemical calculations with semiempirical methods.
|
Implementation of FockMatrixCalculator for DFTB3. It calculates the electronic contributions to the energy and their derivatives with respect to the nuclear cartesian coordinates. More...
#include <ThirdOrderFock.h>

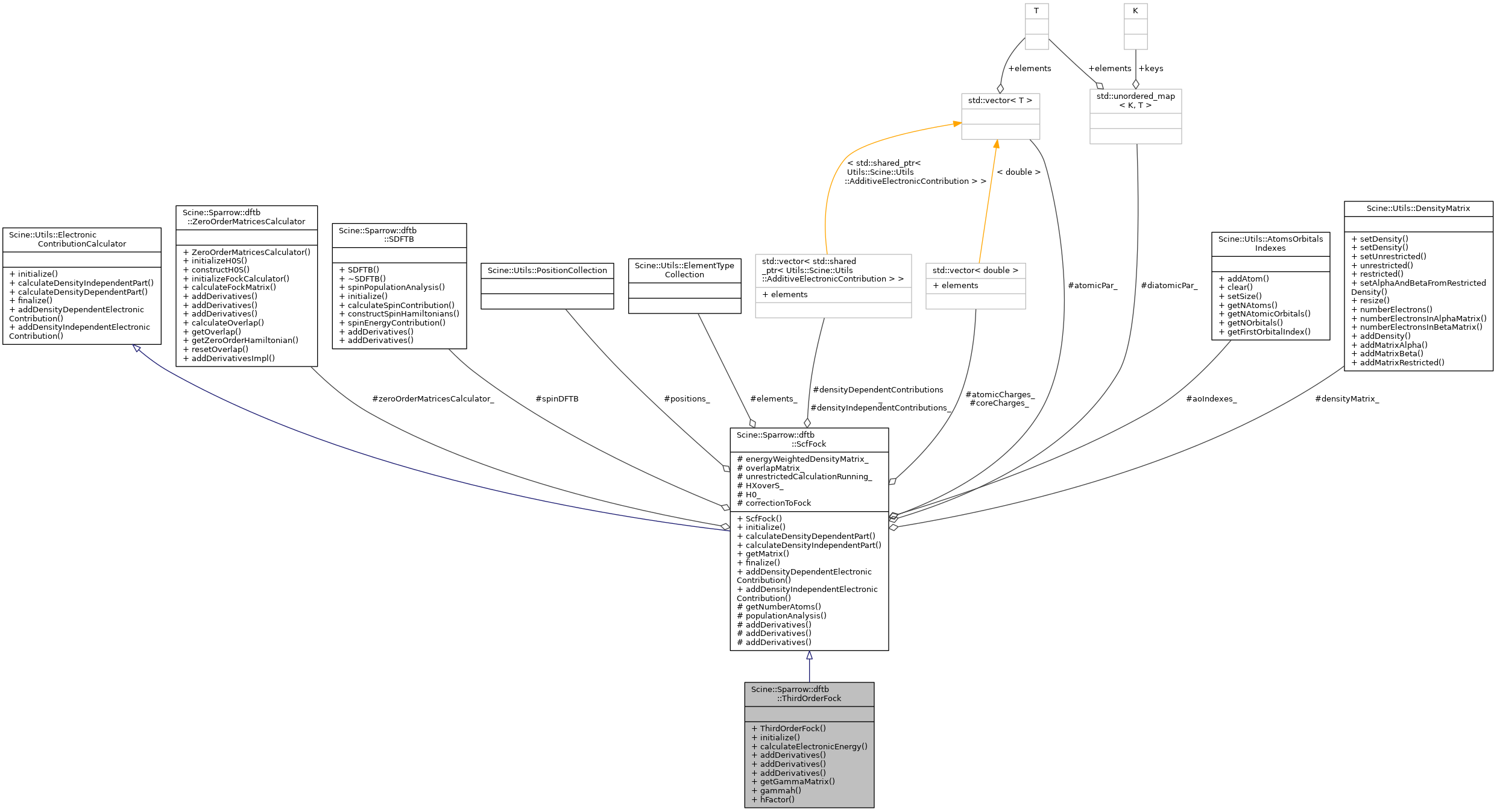
Public Member Functions | |
| ThirdOrderFock (ZeroOrderMatricesCalculator &matricesCalculator, const Utils::ElementTypeCollection &elements, const Utils::PositionCollection &positions, const DFTBCommon::AtomicParameterContainer &atomicPar, const DFTBCommon::DiatomicParameterContainer &diatomicPar, const Utils::DensityMatrix &densityMatrix, const Eigen::MatrixXd &energyWeightedDensityMatrix, std::vector< double > &atomicCharges, const std::vector< double > &coreCharges, const Utils::AtomsOrbitalsIndexes &aoIndexes, const Eigen::MatrixXd &overlapMatrix, const bool &unrestrictedCalculationRunning) | |
| Constructor, calls the ScfFock constructor and sets zeta to 4.0. | |
| void | initialize () override |
| Calls ScfFock::initialize() and initializes the gamma, Gamma and the matrices containing the derivatives. | |
| double | calculateElectronicEnergy () const override |
| Sums up the electronic energy contributions of the zeroth, first and second order Hamiltonian corrections. | |
| void | addDerivatives (Utils::AutomaticDifferentiation::DerivativeContainerType< Utils::Derivative::First > &derivatives) const override |
| calculates automatically the derivatives of the energy with respect to the nuclear cartesian coordinates. | |
| void | addDerivatives (Utils::AutomaticDifferentiation::DerivativeContainerType< Utils::Derivative::SecondAtomic > &derivatives) const override |
| void | addDerivatives (Utils::AutomaticDifferentiation::DerivativeContainerType< Utils::Derivative::SecondFull > &derivatives) const override |
| Eigen::MatrixXd | getGammaMatrix () const |
| template<Utils::DerivativeOrder O> | |
| void | gammah (int a, int b, Value1DType< O > &gamma, Value1DType< O > &Gab, Value1DType< O > &Gba) const |
| template<Utils::DerivativeOrder O> | |
| void | hFactor (double Ua, double Ub, const Value1DType< O > &R, Value1DType< O > &h, Value1DType< O > &dhdU) const |
 Public Member Functions inherited from Scine::Sparrow::dftb::ScfFock Public Member Functions inherited from Scine::Sparrow::dftb::ScfFock | |
| ScfFock (ZeroOrderMatricesCalculator &matricesCalculator, const Utils::ElementTypeCollection &elements, const Utils::PositionCollection &positions, const DFTBCommon::AtomicParameterContainer &atomicPar, const DFTBCommon::DiatomicParameterContainer &diatomicPar, const Utils::DensityMatrix &densityMatrix, const Eigen::MatrixXd &energyWeightedDensityMatrix, std::vector< double > &atomicCharges, const std::vector< double > &coreCharges, const Utils::AtomsOrbitalsIndexes &aoIndexes, const Eigen::MatrixXd &overlapMatrix, const bool &unrestrictedCalculationRunning) | |
| void | calculateDensityDependentPart (Utils::DerivativeOrder order) override |
| void | calculateDensityIndependentPart (Utils::DerivativeOrder order) override |
| Utils::SpinAdaptedMatrix | getMatrix () const override |
| void | finalize (Utils::DerivativeOrder order) override |
| void | addDensityDependentElectronicContribution (std::shared_ptr< Utils::AdditiveElectronicContribution > contribution) final |
| void | addDensityIndependentElectronicContribution (std::shared_ptr< Utils::AdditiveElectronicContribution > contribution) final |
| Public Member Functions inherited from Scine::Utils::ElectronicContributionCalculator | |
| virtual void | addDensityDependentElectronicContribution (std::shared_ptr< AdditiveElectronicContribution > contribution)=0 |
| virtual void | addDensityIndependentElectronicContribution (std::shared_ptr< AdditiveElectronicContribution > contribution)=0 |
Additional Inherited Members | |
| Protected Member Functions inherited from Scine::Sparrow::dftb::ScfFock | |
| int | getNumberAtoms () const |
| void | populationAnalysis () |
| void | addDerivatives (Utils::AutomaticDifferentiation::DerivativeContainerType< Utils::Derivative::First > &derivatives) const override |
| adds the derivatives for the first, second atomic and second full types. | |
| void | addDerivatives (Utils::AutomaticDifferentiation::DerivativeContainerType< Utils::Derivative::SecondAtomic > &derivatives) const override |
| void | addDerivatives (Utils::AutomaticDifferentiation::DerivativeContainerType< Utils::Derivative::SecondFull > &derivatives) const override |
| Protected Attributes inherited from Scine::Sparrow::dftb::ScfFock | |
| ZeroOrderMatricesCalculator & | zeroOrderMatricesCalculator_ |
|
const Utils::ElementTypeCollection & | elements_ |
| const Utils::PositionCollection & | positions_ |
|
const DFTBCommon::AtomicParameterContainer & | atomicPar_ |
|
const DFTBCommon::DiatomicParameterContainer & | diatomicPar_ |
| const Utils::DensityMatrix & | densityMatrix_ |
| const Eigen::MatrixXd & | energyWeightedDensityMatrix_ |
| std::vector< double > & | atomicCharges_ |
| const std::vector< double > & | coreCharges_ |
| const Utils::AtomsOrbitalsIndexes & | aoIndexes_ |
| const Eigen::MatrixXd & | overlapMatrix_ |
| const bool & | unrestrictedCalculationRunning_ |
| SDFTB | spinDFTB |
| Eigen::MatrixXd | HXoverS_ |
| Eigen::MatrixXd | H0_ |
| Eigen::MatrixXd | correctionToFock |
|
std::vector< std::shared_ptr < Utils::AdditiveElectronicContribution > > | densityDependentContributions_ |
|
std::vector< std::shared_ptr < Utils::AdditiveElectronicContribution > > | densityIndependentContributions_ |
Implementation of FockMatrixCalculator for DFTB3. It calculates the electronic contributions to the energy and their derivatives with respect to the nuclear cartesian coordinates.
 1.8.5
1.8.5 
